The plain-English version
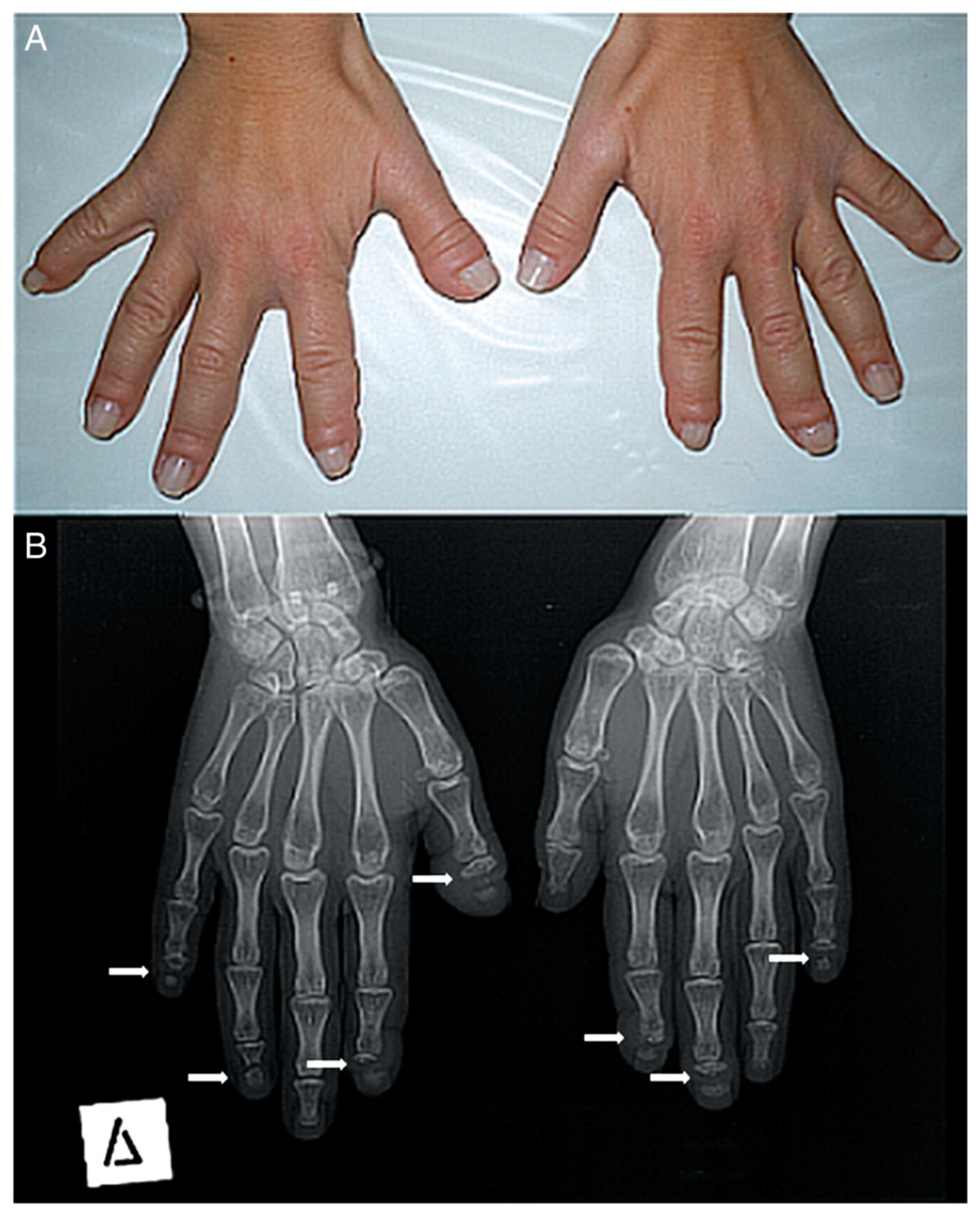
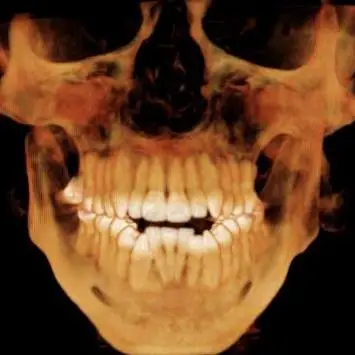
Hajdu-Cheney Syndrome — pronounced "hay-dew chaye-knee" — is a rare bone and connective-tissue disorder. Your skeleton is doing two jobs at the same time, every day of your life: building bone and breaking bone down. In HCS the breakdown crews run faster than the builder crews. The result is bones that get thinner, smaller, and less stable over time, especially in the hands and feet, where the tips of the fingers and toes can resorb back into the body. The connective tissues are still quite the mystery, and I am not sure how exactly they are affected by the mutated gene. The medical word for that bone loss and clubbing at the tips is acro-osteolysis. It is a headline symptom but not the only one — HCS reaches into the spine, the skull, the jaw, the heart, the kidneys, and the immune system. The longer you have it, the more of you it gets to. Some of us start out a lot further along than others who barely have symptoms until much later in life. I am fortunate mine is relatively tame.
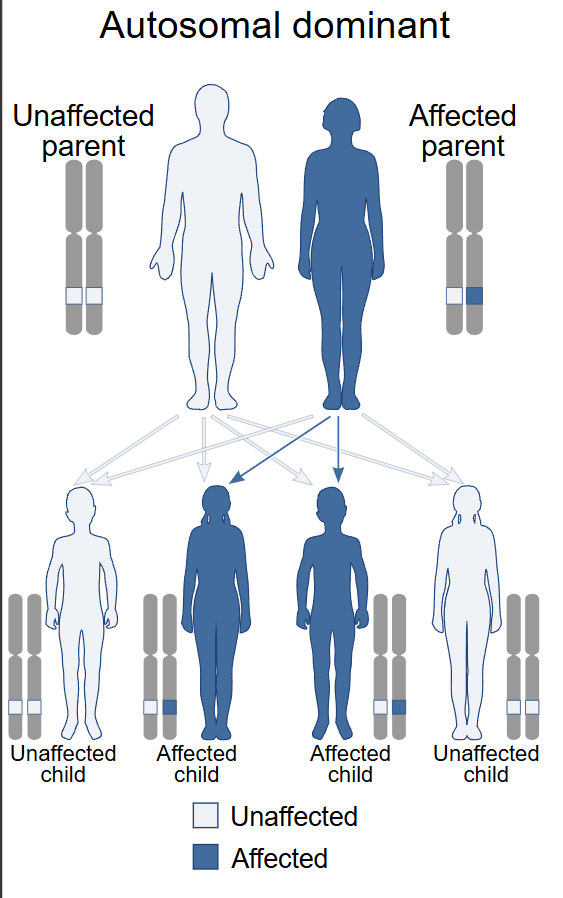
It is autosomal dominant, which is the polite way of saying one parent passes it on to roughly half their children. Many cases also appear out of nowhere — they call it de novo, a new spontaneous mutation, no family history. There are roughly a hundred documented cases in the medical literature and not even fifty of us alive in the world right now, give or take. I base this only off of our Facebook group which is the only place we seem to have congregated. I scan other social medias once in awhile out of curiosity, but it has been crickets.
That last sentence took me twenty years to be able to write without flinching.
My version of it
I was diagnosed at three or four years old. The first thing my mother noticed was a thumb that wasn't shaped quite right. I remember my dad closing the door on my thumb a couple times on accident when I stuck my hand in it. Kids. Over the next forty years that one thumb became both hands, my wrists that wouldn't bend backwards, then the shoulders that wouldn't go above my head, then the feet that flattened, then the neck that fractured and dislocated.
My case is unusual in one important way. I have been tested twice for the known NOTCH2 mutations that cause HCS in most people, and both times the test came back clean. I do not carry a recognized pathogenic variant. My diagnosis is clinical — confirmed on the way my body has built itself, not on a line of code in my genome. That makes me a footnote in my own disease. Most published case studies are children or middle-aged adults with a confirmed mutation; I am forty-five, with the phenotype of a textbook case and the genotype of someone the textbook hasn't met yet.
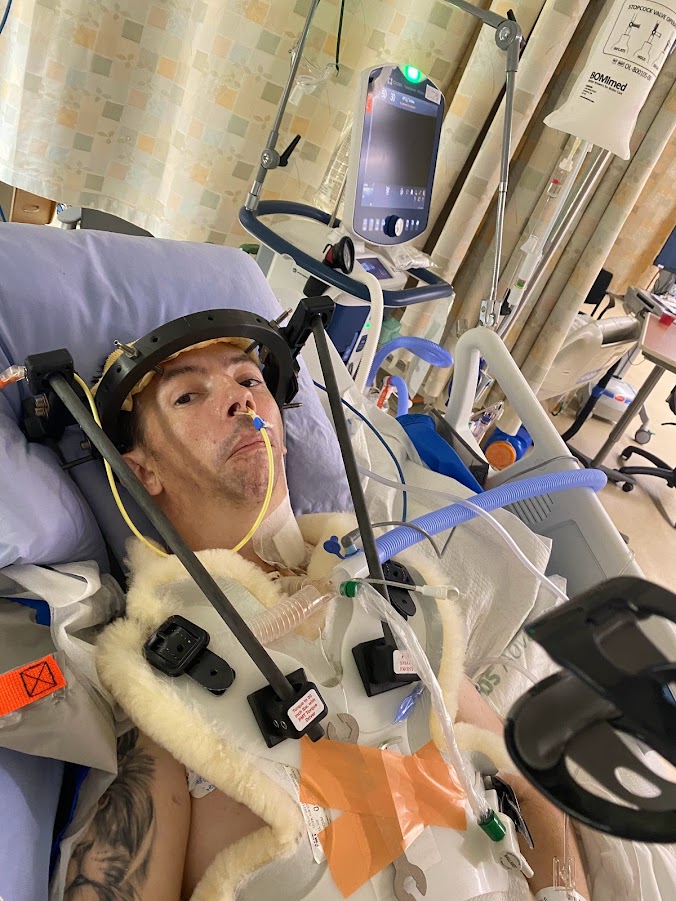
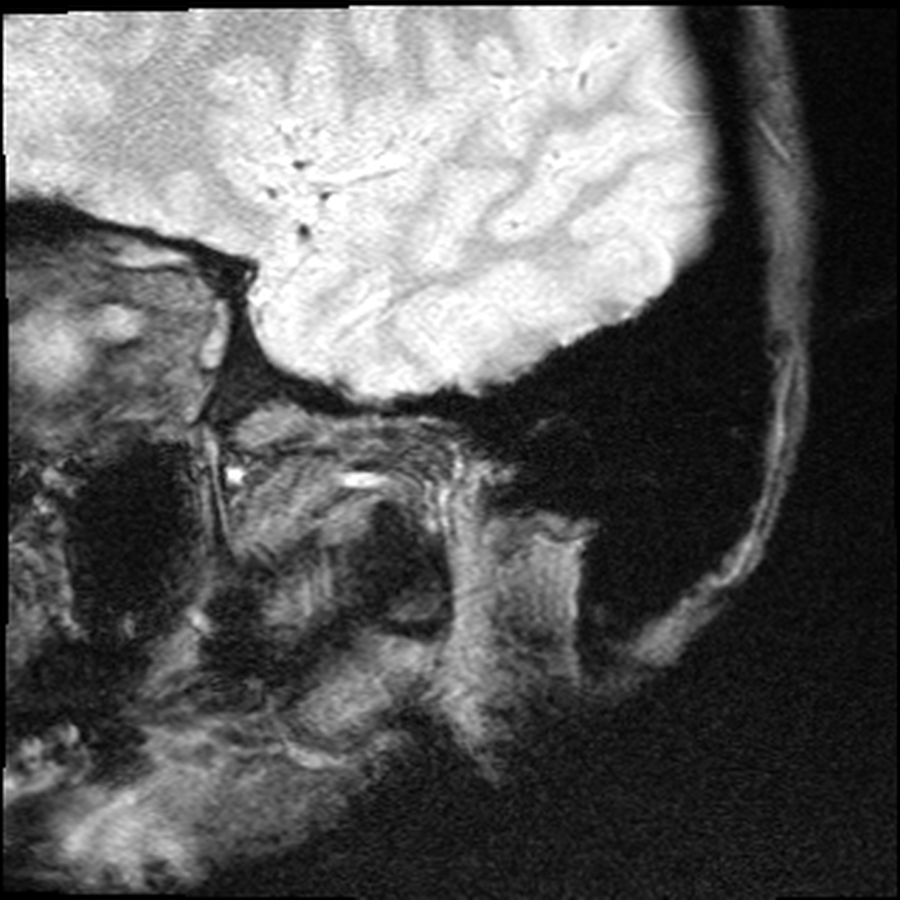
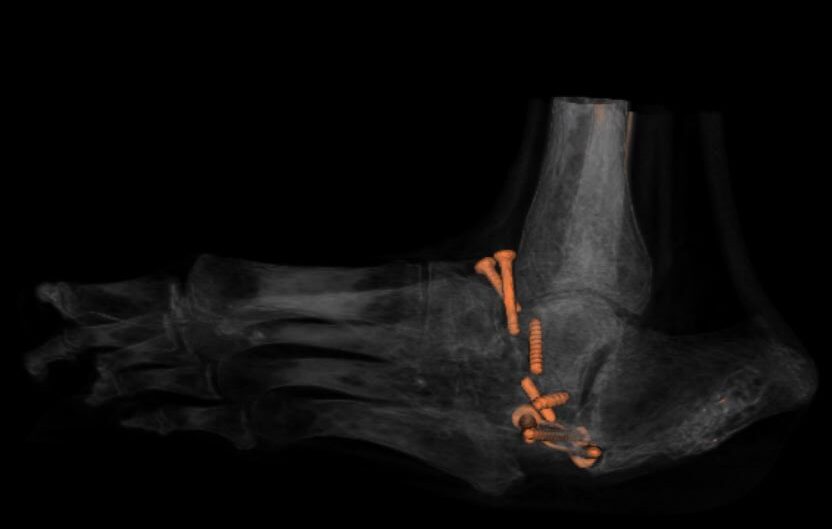
My version, in fast-forward: childhood pneumonias every other year, growing up on farms doing the manual labour the other kids did. A neck injury before I can remember limited my range of motion turning my head. I was injured once at a chiropractor. A basketball injury at fifteen that knocked my neck into permanent slightly more reduced range. A culinary career — The Keg, SAIT, then Ric's Grill, then Township 71, then teaching at GPRC, then Majors Homestyle. Twenty years of bisphosphonates starting in 2004. A neck DVT in 2017 because I have no clavicles and pinched a vein closed in my sleep. A sledding accident in January 2022 that nearly finished off what was left of the alignment in my upper spine. A couple accidents playing with kids finished it off leading to a cervical fusion in July 2022 that runs from the base of my skull to my T3 — three rods, twelve plates, forty-odd screws, donor bone, and enough bone glue for three people. I weigh 98 pounds today. My BMI is 13.6. The right condyle of my jaw is gone; the left is partially eroded.
If you've read my chronological story on Bare Your Rare, you already have the timeline. This page isn't the timeline. This page is what the timeline turned out to mean instead.
The thing I wish someone had told me at ten: every HCS case is its own dialect of the disease. The textbook traits are a menu, not a destination. You will probably get some of them and not others, in an order nobody can predict. That uncertainty is the part that nobody writes about because it doesn't sound clinical enough to publish. There are some trends noticed recently such as the upper airway infections frequently in younger years, organ issues later in life.
What I lost

The kitchen went first, and the kitchen was where I lived. I started cooking in 2001 to pay my way through the last year of college at The Keg and I would go on to spend the next 12 years cooking and running the kitchen. I worked my way up the line over 2 years and then became assistant kitchen manager, then kitchen manager for 10 years. In 2011 I began my schooling for the apprenticeship at SAIT. I finished in the fall of 2012. Eventually I had to move on to try a different angle and that's when I started as Head Chef at Ric's Grill the day Patience was born, ten days late.
I ran Township 71 after shutting down Ric's Grill. At the same time all this was going on I taught the cooking part of the Hospitality and Tourism Diploma at the college up here in Grande Prairie. The last full kitchen I ran was Majors Homestyle and Tractor Jack's. By 2018 I couldn't sustain eight let alone twelve hours on the line with a thirty-pound stockpot and a 50lb box of potatoes. Standing and walking twelve miles a day across an eight-to-sixteen-hour shift was no joy. I knew before everyone around me that the kitchen was going to lose this argument. I'm grateful I left on my own terms. I am also still angry about it some days, I felt like I had years left in me and I did not intend to let my family down the way I did. I should have left years before so that I would have the gas left for my new family. Even today I am torn between not wanting to overdo it and at the same time there is a lot of guilt for not helping more with housework and chores. I wish to rest my feet as much as possible because no matter what I do they ache every day. I wake at 5am and they ache, I think about going to Walmart or the Superstore or the Mall and they yell “NOOOOOOOO!” at me before we even go.
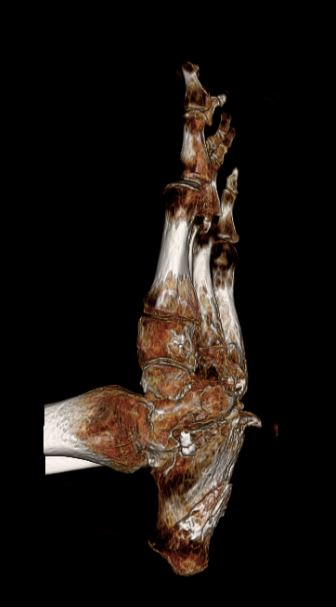
The hands took the kitchen. After the hands, slower losses, the kind that don't have one moment to point at. Both wrists that won't bend backwards. Shoulders that won't lift above my head. A left thumb dislocated in a few places. A right hand more degenerated than the left, but the left compensated by losing a ring finger to partial dislocation. Most of my fingertips are dissolved or gone. There were bone spikes growing on the bottom of my feet. Both feet have very little up-and-down rocking left. The arches are inverted.

The current ledger, since you asked. My ribs hurt — the right back ribs especially, where they don't unite cleanly anymore, and the front ones that have started to feel like they're pressing in on the lung. That is not a cool feeling. My back hurts at T8/9, where there's a wedge fracture and a big osteophyte that doesn't care what chair I'm sitting in. My right shoulder is in a queue for an ultrasound to be booked; I've been waiting for that call since mid-February. The bone scan I'm waiting for has been in the same queue since the same week. I need iron levels checked, a fluoroscopy, a colonoscopy. I have nocturnal bowel movements I'm not used to having, and the occasional accident I'm even less used to. The ribs and the back wake me up at night. I tried six weeks of low-FODMAP and got about ten percent of my gut back, which is something but is not enough to claim victory. I had a cortisone shot in a finger that made the pain worse for two weeks; the same finger turned out to have an infection in the tip; the nail was surgically cut off; two rounds of antibiotics; the nail is growing back; it still bothers me. I am thinking about going back on oxycodone, which is the kind of decision that is difficult to make, seldom is there a way back when one is degenerative like this. I think it is common when someone's back is in rough shape.
That is what HCS is on a Tuesday afternoon as I am writing this on.
What it gave
The reframe in the subtitle is not glib. I had to earn it.
The biggest thing HCS gave me is time. Not extra time — there is no extra of anything when your spine is fused and your weight is dropping. Time back. The hours a chef spends on a line are not yours. They are the kitchen's. When the kitchen took itself off the table, those hours came back. I have used them poorly some days and well most days, and the well days have produced more than I would have predicted.

It gave me my kids. Melanie and I had been apart for a decade when we got back together, and we had three kids in five years — Daniel, Patience, and Faith. The man who could still chef sixteen-hour shifts would not have been the father these three kids needed. The man whose body grounded him at home is. That is a hard sentence to type without it sounding like a cope. It is not a cope. It is the truth I have arrived at after living with it.
It gave me a mind for the literature. When you have a disease that fewer than a hundred people are documented with, you stop waiting for someone to explain it to you. You read the papers. You learn what NOTCH2 is, what FBW7 does, what the PEST domain is, what it means that yours got truncated. You become — not a doctor — but a reader of doctors, fluent enough to ask the right questions and to flag the answers that don't add up.
It gave me three websites, which is funny to type given that I'm not a programmer. This one is the personal hub. Bare Your Rare is the writing project about HCS specifically and rare diseases in general. The GP Residential Society is the volunteer board I sit on. I work with Claude to build them — I don't write the code; I spec the design, the voice, the editorial moves, and Claude writes them out. Grok and ChatGPT also teach me along the way and handle the non-coding such as image generation, some of the diagrams, research, and 2nd or even 3rd opinions. Three sites in eighteen months says something about how that collaboration goes. I think I have more than 22,000 prompts not counting ChatGPT. I have become an AI wiz and my next project will be an app — a game perhaps.
Back to the story… It gave me the family record. Since I left the kitchen I have sporadically used my time putting together the family history — all the way back to the 1600s on a couple branches; my own thread through all of this is the long way round. The long-form research goes on YouTube as documentaries. The condensed versions live in the heritage section of this site. I don't know if anyone will read it but it is here. Perhaps one of my kids will take a trip on the way-back-machine and fill a desire to learn where they came from. Our families are older, fragile, and smaller.
I am not going to tell you HCS is a gift. It is not. But it has, in the way that constraints often do, drawn the lines around the life I now have, and the life inside those lines is one I would not trade. That is the closest I can get to honest.
The genetic question

When Melanie and I decided to have children, we knew the math. HCS is autosomal dominant — fifty percent. Each pregnancy was a coin flip, and we flipped it three times, knowing.
I will not pretend that decision was easy or that it should be easy for anyone. Autosomal-dominant rare disease is one of the few medical situations where what you know about yourself becomes a load-bearing input into who else gets to exist. The honest argument I sat with for years was this: the same condition that has made my life harder has also made it richer in ways I cannot subtract from. If I wouldn't trade my life for a HCS-free version of it, on what grounds was I supposed to refuse my children theirs?
We have three kids. None of them are showing early phenotype features. What I will say is that I watch them more closely than I would have otherwise, that their pediatricians and the family doctor know what to look for, and that the tools that were not available when I was diagnosed in 1984 are available to them now. We will not do genetic testing on the children, I will not be responsible for their DNA being used in any way that can be stolen or lost or sold.
If you are facing this decision yourself: I will not tell you what to do. I will tell you that the calculus is not "spare them the disease," because the disease is not the only thing they would inherit. The calculus is, "what kind of life will the person I would be raising them want to have lived." That is the only honest version of the question.
What I wish I had known
A lot of advice for the newly diagnosed comes from people who haven't been at it for forty years. Here is what I have learned from forty years of being at it.
Apply for the disability program the day your earning capacity drops, not the day you can't physically work. In Alberta that program is AISH. The medical paperwork — Part B in our case — takes weeks and if it is rejected it can take months to contest. Income Support is the bridge while you wait. I made the mistake of waiting too long. The bridge was harder to build than it needed to be. It was harder on my employer and coworkers.
Photograph and back up every page of your medical paperwork. The same documents will be asked for again at every appeal, every program transition, every specialist intake. The first copy you make is the easiest. Make three. If you worked there is your disability pension, provincial/state benefits, some municipal benefits though they only require income statements. If you have a rare condition and do your own research your files are very helpful.
CPP Disability and AISH stack, but AISH claws back the CPPD dollar-for-dollar. Apply to both. Understand that getting CPPD does not raise your income — it just shifts which level of government is paying you. Knowing that in advance saves a lot of "did I do something wrong" feelings on the first deposit day after the change.
The cohabiting-partner rule will quietly cost you money. Before you move in with a partner — or get married — request a benefit-impact estimate in writing from your case worker. Don't learn this from next month's deposit.
Keep a paper file of every Notice of Decision. The twelve-month re-assessment window starts from the date on that letter. After that it is a brand-new application.
The "rapid reinstatement" rule is your safety net for trying work. If you leave AISH for employment and earnings later drop, you can be reinstated within two years without a fresh medical. Knowing that exists makes attempting work less terrifying. Most people don't know it exists.
Sleep is a metric. If your back and ribs are waking you at night, that is not a "tough it out" problem. That is a planning problem and a medication problem and a sometimes-an-imaging problem. Track your nights. Bring the count to the appointment.
Your body is a teaching text and that is fine. I tend to be unusual in clinical settings. There was a whole nursing class that came by to watch them put my halo on. I don't mind. The next person walking into a halo brace will get more careful hands because of those students. If you are willing to be the example, say so. If you are not, say that too.
The research detour
This is the part of the page nobody else can write about my disease, because nobody else has been reading my disease from the inside for forty years.
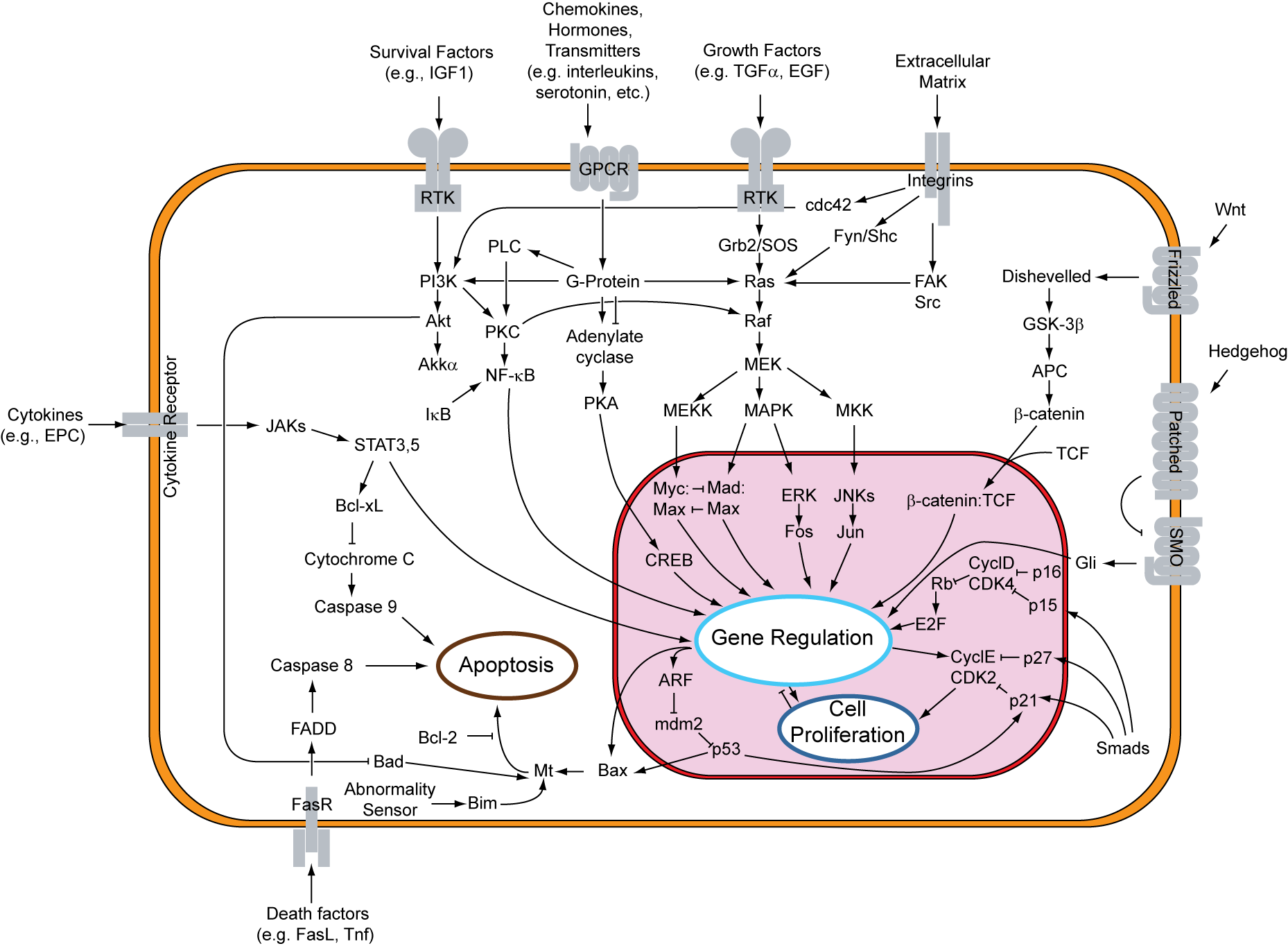
HCS is caused — for the people who carry the recognized version — by a single faulty copy of a gene called NOTCH2. NOTCH2 sits on chromosome 1, and it codes for a kind of antenna that pokes out of cells, ready to receive a signal from the cell next door. When the signal arrives, the antenna gets snipped, the snipped piece travels to the nucleus, and the cell decides what to do next — what to become, when to divide, when to die. This is the Notch signaling pathway, and it is one of the oldest tools evolution has. Fruit flies use it. Worms use it. We use it every day, in development and in adulthood.
The HCS-causing mutations are clustered in the very last exon of NOTCH2. (I don't know if 2 of us have the same exact mutation, some intergenerational afflicted families should have the same mutation though.) They cut the protein short, and the piece that gets cut off is called the PEST domain — proline-glutamate-serine-threonine, hence P-E-S-T. (Just don't ask why the glutamate = E, it is a mystery to me) The PEST tail is the cell's expiry sticker. It tells the cleanup machinery: when this protein has done its job, throw it out. Without the sticker, the protein is no longer flagged for removal. It lingers. The signal stays on too long. In bone, that means more osteoclasts — bone-breakdown cells — and not enough rebuild. The skeleton tilts toward demolition. The demolition crew runs without oversight — a metaphor I picked up from the literature and now think about most days.
The cleanup enzyme that normally tags the PEST tail is called FBW7. It is one of about six hundred enzymes called E3 ubiquitin ligases — molecular sticker-guns. When researchers knocked out FBW7 specifically in mouse osteoclasts, the mice developed an HCS-like skeleton. When they treated those mice with a Notch inhibitor, the bone loss reversed. That is a clean proof of mechanism, and it is one of the reasons I am cautiously optimistic about the next generation of treatments.
NOTCH2's main partner ligand is JAG1 — Jagged1 — and JAG1 is the bridge between bone disease and blood disease. In bone marrow, Jagged1 helps decide whether stem cells stay quiet, divide, or commit to becoming red blood cells, white cells, or platelets. The same axis that is dialed too loud in my skeleton is also instructing my immune system. Most of us with HCS don't show dramatic blood phenotypes — but the link is there, and it is one reason I read my own bloodwork carefully.
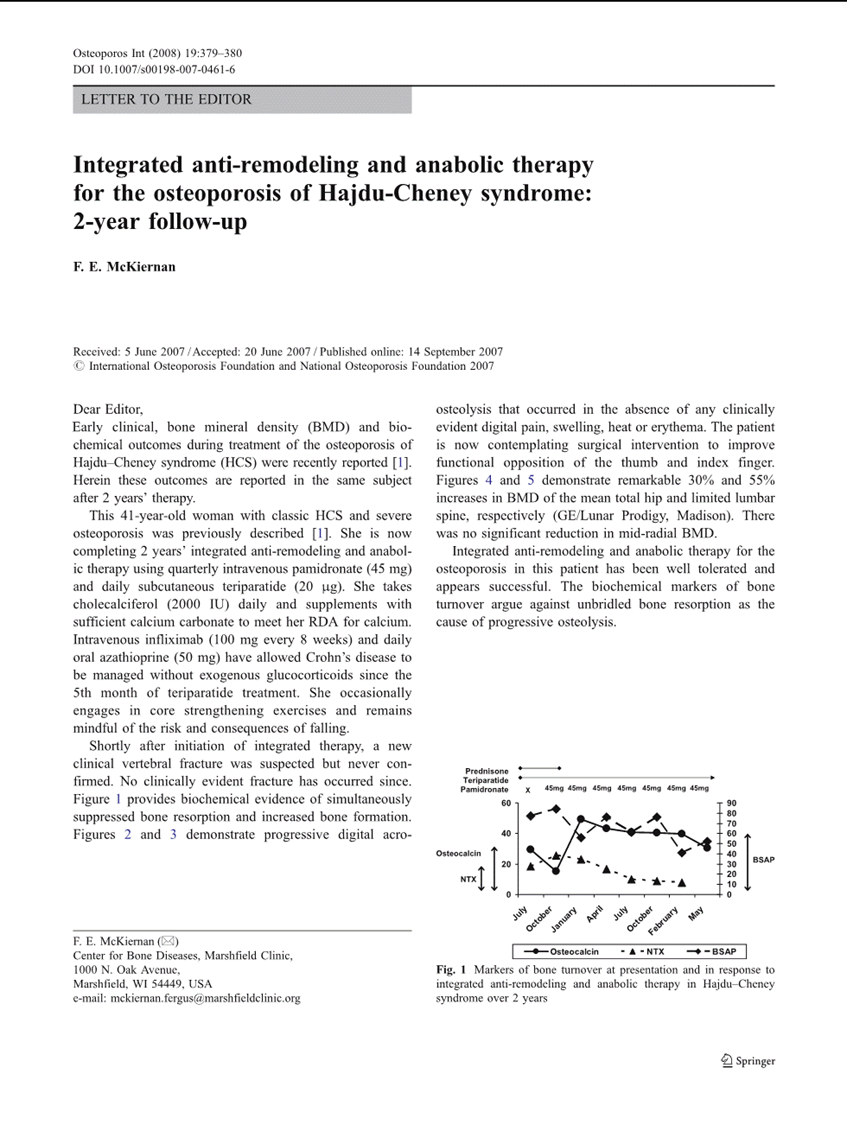
Where does that leave treatment? Bisphosphonates — pamidronate, then alendronate, then zoledronic acid for me — work downstream by poisoning osteoclasts directly. They do not fix the Notch problem; they slow the damage. Denosumab blocks RANKL, which is the activator the osteoclasts respond to; it raises bone density without stopping the acro-osteolysis itself. Romosozumab — Evenity — is the new option I am tracking; it builds bone instead of just slowing breakdown. It is not approved for HCS in Canada (it is on-label for postmenopausal osteoporosis), but the mechanism is right and the trial data are good. Anti-Notch antibodies are the frontier; they are not in HCS trials yet, but they are in the literature, and the mouse data are encouraging.
I read the literature because nobody else is going to read it for me, and because the scale of the disease — not even fifty of us alive — means that the patients themselves are most of the dataset. I am Care4Rare-enrolled. I send my imaging when researchers ask. I keep my own files cleaner than my own desk. I think of myself as one row in a registry that does not yet exist at the size it would need to be.
If you read one paper, read Elizabeth Bombal's 2022 Master's thesis — Assessment and Development of Educational Resources for Hajdu-Cheney Syndrome. Elizabeth has HCS herself. The thesis is the most useful single document on what it is like to live with this disease and what the research community is and is not doing for us. It is in the resources list at the bottom of this page.
What I write about now
After forty years of HCS, I had a choice about what to do with the literacy it had built in me. I built a website for it.
Bare Your Rare is a writing project for HCS specifically and for rare disease in general. It is the home of the work that does not fit on a personal page. Two pieces in particular complement what you have just read here:
- Thomas's HCS Story — the chronological, year-by-year version of my life with HCS. This page you're reading is the essay; that page is the timeline. Both are true; they answer different questions.
- The HCS Patient Guide — what to do about HCS. Which specialists to assemble. What to monitor. What to flag in an emergency room. What surgeons need to know before they touch you. If you or someone you love has HCS, that is the document I wish someone had handed me at twenty-two.
The Resources section of Bare Your Rare also covers the broader rare-disease landscape — the AI tools that are starting to make ultra-rare conditions diagnosable, the registries worth joining, the patterns that show up across rare diseases as a class. None of that fits here. All of it is one click away.
Common questions
The short answers, for anyone who landed here looking for them. The longer story is everything above.
What is Hajdu-Cheney Syndrome?
A rare bone and connective-tissue disorder. Your skeleton is always doing two jobs at once — building bone and breaking it down — and in HCS the breakdown runs faster than the rebuild. Bones get thinner, smaller, and less stable over time, especially in the hands and feet, where the tips of the fingers and toes can resorb back into the body. It can also reach the spine, skull, jaw, heart, kidneys, and immune system; the longer you have it, the more of you it tends to reach.
How do you pronounce “Hajdu-Cheney”?
Hay-dew chaye-knee.
What is acro-osteolysis?
It is the medical word for the bone loss and clubbing at the tips of the fingers and toes — the bone literally dissolving back. It is the headline symptom of HCS, but not the only one.
How rare is it? How many people have it?
There are roughly a hundred documented cases in the medical literature, and — going by our Facebook group, the one place we seem to have congregated — likely fewer than fifty of us alive in the world right now, give or take.
Is it inherited?
It is autosomal dominant, which means a parent passes it to roughly half their children — a coin flip per pregnancy. But many cases appear out of nowhere too: a new, spontaneous mutation (de novo) with no family history.
What causes it — what gene is involved?
For most people who carry the recognized form, a single faulty copy of a gene called NOTCH2. The HCS mutations cluster in the gene’s last exon and cut the protein short, removing the “expiry sticker” (the PEST domain) that normally tells the body to clear it — so the bone-breakdown signal stays switched on too long. (My own genetic tests came back clean twice; my diagnosis is clinical.)
Can it be treated? Is there a cure?
There is no cure yet. Treatment slows the damage: bisphosphonates (pamidronate, alendronate, zoledronic acid) and denosumab reduce bone breakdown, and romosozumab (Evenity) builds bone and is promising, though it is not approved for HCS in Canada. Anti-Notch antibodies are the research frontier.
What symptoms does it cause?
It varies enormously — every case is its own dialect of the disease. Common features include the fingertip and toe bone loss (acro-osteolysis), low body weight, a spine and skull that change shape over time, jaw (TMJ) erosion, frequent upper-airway infections in the younger years, and organ involvement later in life. The textbook traits are a menu, not a destination.
I think I, or someone I love, might have HCS — where do I start?
You are not alone, even though it can feel that way. The HCS Patient Guide and my own story on Bare Your Rare are the documents I wish someone had handed me; the OMIM and GeneReviews entries in the Resources below are the authoritative medical references; and registries like Care4Rare and NORD are worth joining. You are also welcome to email me.
Resources
Curated. Not exhaustive. The resources I have actually used, in roughly the order I would point a newly-diagnosed family member to them.
For HCS specifically
- Case Studies & Research — my curated library of the free HCS case reports, NOTCH2 genetic research, and RANK-L studies worth reading.
- Bare Your Rare — my own writing on HCS, including the patient guide, my chronological story, and the Resources page that goes deeper than this one.
- Elizabeth Bombal's 2022 thesis, Assessment and Development of Educational Resources for Hajdu-Cheney Syndrome — the most useful single document by a fellow patient. Available through the University of Connecticut.
- OMIM #102500 — the Online Mendelian Inheritance in Man entry for HCS. Authoritative gene-and-phenotype reference.
- GeneReviews — NOTCH2-related Hajdu-Cheney Syndrome — a clinically-oriented overview, updated periodically.
- Orphanet — HCS — the European rare-disease portal's HCS entry; useful prevalence and natural-history data.
Patient registries and research networks
- NORD — National Organization for Rare Disorders and the IAMRARE registry platform.
- Care4Rare Canada — the Canadian rare-disease research consortium.
- CORD — Canadian Organization for Rare Disorders — Canadian rare-disease policy and advocacy.
- RareConnect — moderated patient communities by condition. The HCS group is small but real.
- Sanford CoRDS — registry program covering hundreds of rare conditions, HCS included.
- EURORDIS — European patient organization; their resources translate well across borders.
- Rare Diseases Clinical Research Network (RDCRN) — NIH-funded consortium running the long-form natural history studies HCS doesn't yet have but might one day get.
- Global Genes — patient advocacy infrastructure; their toolkits are good.
For the science
- NIH GARD — Genetic and Rare Diseases Information Center.
- ClinVar and VarSome — variant databases. If you have a NOTCH2 variant, this is where you check what is known about it.
- The 2011 Nature Genetics papers — the original NOTCH2-as-cause-of-HCS papers, Simpson et al. and Isidor et al. Behind paywalls but the abstracts are free and the citation chain leads everywhere else.
Canadian disability navigation
- Alberta AISH — the program itself.
- CPP Disability — federal program that stacks with AISH.
- Disability Tax Credit — apply once, helps every year.
- The Canada Disability Benefit (CDB).
- The forthcoming Alberta Disability Assistance Program (ADAP) — for those who can do some work; replaces parts of the AISH structure.
If you have a resource I should add, send it to me.

There are a hundred of us documented in the medical literature. Less than fifty of us are still alive. If you are one of us, or you love one of us, or you treat one of us, or you've stumbled onto this page because something in your body is acting like something on this page — please know that you are not alone, and please consider reaching out — and if you'd rather find others who are actually living it, that's what I built Bare Your Rare for.
The fastest way to reach me is email: gubznfzpurrfzna@tznvy.pbz
Thomas Cheesman
Grande Prairie, Alberta
April 2026